
Alex Tang
Articles
- General
- Theology
- Paul
- Karl Barth
- Spiritual Formation
- Christian Education
- Spiritual Direction
- Spirituality
- Worship
- Church
- Parenting
- Medical
- Bioethics
- Books Reviews
- Videos
- Audios
- PhD dissertation
Spiritual writing
- e-Reflections
- Devotions
- The Abba Ah Beng Chronicles
- Bible Lands
- Conversations with my granddaughter
- Conversations with my grandson
- Poems
- Prayers
Nurturing/ Teaching Courses
- Sermons
- Beginning Christian Life Studies
- The Apostles' Creed
- Child Health and Nutrition
- Biomedical Ethics
- Spiritual Direction
- Spiritual Formation
- Spiritual formation communities
- Retreats
Engaging Culture
- Bioethics
- Glocalisation
- Books and Reading
- A Writing Life
- Star Trek
- Science Fiction
- Comics
- Movies
- Gaming
- Photography
- The End is Near
My Notebook
My blogs
- Spiritual Formation on the Run
- Random Musings from a Doctor's Chair
- Random Sermons from a Doctor's Chair
- Random Writings from a Doctor's Chair
- Random Spirituality from a Doctor's Chair
Books Recommendation
---------------------
Medical Students /Paediatric notes
|
|
|||
|
Kairos Spiritual Formation Ministries
Podcast
Tang's Takeaways
Read more //alextang.substack.com/p/engaging-dragons
Courses 2024
Courses 2023
Micro Learning Course
Webinar
Sermons
More sermons on my
Free Books Download
Guidance for Churches in the Coronavirus Era
Citation: Amar-Singh HSS, Lim Swee-Im, Matthew Ling Ung-Hiing, Alex Tang Tuck-Hon, Low Chai-Hok, David Bok. 2020. Guidance for Churches in the Coronavirus Era. Malaysia. Version 1, 2nd May 2020. Creative Commons Attribution CC BY 4.0
free download here
Panduan untuk Gereja Dalam Era Coronavirus
free download here
free download here Posts Lifehacks from Eugene Peterson The Spirituality of Eugene Peterson: Seven Lifehacks for the Spiritual Life.
The Metaverse is Web 3.0, the next step in the development of the Internet. The evolution of the Internet as a private data sharing network for scientists with its clumsy modem to its user-friendly browser user-friendly interface has been very rapid. Web 1.0 is when the webpages are static and we can only read off them. We cannot interact with them by adding or subtracting. Then came Web 2.0 which was a marvellous interactive experience. We can edit, produce, and chat using that technology. There was a proliferation of chat groups that lead to blogs, personal websites, add sound and video, and social media such as Facebook and Twitter. Web 2.0 for all its benefits is still 2-D. It still remains on the screen. Web 3.0 or Metaverse is 3-D. Content with which we can interact is no longer flat. It is now 3-dimensional. The movie Ready Player One is a good visualization of what Metaverse is. In the first half of this article, I will describe what Metaverse is and then I will share some implications this will have on Christians and the Church. Read more | More related articles in Glocalisation
conversation with my avatar in spatial.io
closing prayer in a Metaverse church
download Koinonia Paper 02/2022 The Gift of Listening
The Gift of Listening: Spiritual Direction. Webinar organised by KLCentre, Seminari Theologi Malaysia (STM) on 24 August 2021. Fundamentals of soul care, listening, discernment and spiritual direction.
Christian Considerations on COVID-19 Vaccines
Christians and Christian faith communities are facing a new dilemma. Should they and the families receive the COVID-19 vaccination? This is not about being anti-vax or vaccine resister. This is about making a choice. read more
Discerning the Calling in the Marketplace Webinar
Reimagining Christian Spiritualities in this COVID-19 Pandemic Webinar
Webinar for healthcare professionals by Medical Dental Christian Fellowship Malaysia and supported by ICMDA read more more Spirituality Guidance for Churches in Post-COVID-19 Era Webinar
Moving Beyond Lament Webinar
Dispatches from the COVID-19 Front The Borders are Closed and We are in Lockdown Don’t Travel, Stay at Home Telling the Truth Do Nor Covid Your Neighbor Stop, Pause, Reset One Anothering in COVID Lockdown For Times Such As This A Strong Spirit in Face of Adversity Anxiety is Emotional and Spiritual Bondage Covid Anger It Ain't Over Till It's Over Along Came a Donkey Them and Us Lamentation during the Pandemic Navigating Stormy Seas Christian Considerations on COVID-19 Vaccines Christmas is not Cancelled in COVID-19 Pandemic
read the dispatches at my blog www.draltang.wordpress.com
Contemplative Spirituality Series
A contemplative life is the foundational basis of our spirituality. more Spiritual Formation Spirituality
There is a state of fear in people of many countries which are far from the epicentre of the 2019 nCoV epidemic in Wuhan and Hebei provincial, China. Even though there are only a handful of patients in their countries, and most of these patients have been to the epicentre, or being in contact with people from there, the state of fear of being infected by the coronavirus is extremely high. read more more articles on Medical
Do we have spiritual senses that mirror our physical senses? read more more Spirituality
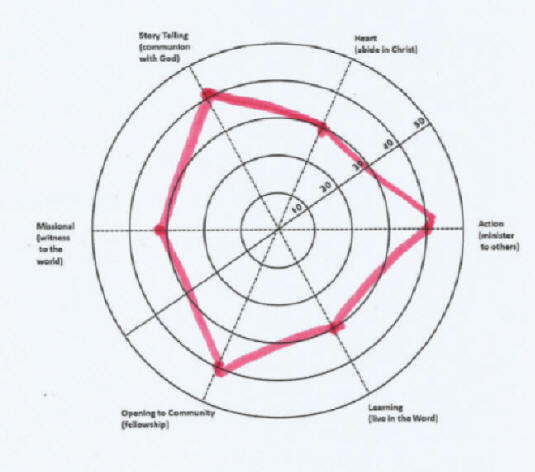
Spiritual formation inventory is a spiritual assessment tool to obtain a snapshot of our spiritual life at a certain moment in our spiritual journey. This tool will help us to discover what areas in our spiritual practices are strong and what the weaknesses are. For effective spiritual formation or spiritual growth, a balanced spiritual life and practices is essential. Too much emphasis on one aspect of our spiritual practices at the expense of others may lead to an unhealthy spirituality. read more more Spiritual Formation
The Ethics of Genome Editing: A Christian Perspective
We need a framework to look at the rapidly advancing challenges of emerging new technologies. Technologies such a genome science, Big Data, Artificial Intelligence, and the Digital Person will redefine the structure and nature of our civilization within the next few years. Are these technology helpful or harmful? What should be the Christian faith communities’ respond to them? These new technologies would not be found in the Bible, a text that was written more than two thousand years ago. Where then are Christian to seek guidance for their discernment? A framework to guide our thinking is needed.
read more
more Bioethics
Published in Biblical Graduate School of Theology (BGST), Singapore March 2019 Newsletter
read more more Spirituality
Artificial Intelligence and God
The subject of artificial intelligence is an area of concern and even fear among computer scientists, sociologists and theologians in the last couple of years. Artificial intelligence as self-learning and self-improving software has beaten chess grandmasters and recently human computer gamers in a complex online game named Starcraft II. The AI software was actually learning from both its successes and mistakes. Initially called machine learning, now it has a better name of deep learning. read more more Bioethics
Safespace Interview on Biotechnology: Are We Playing God?
Biotechnology is both a blessing and a curse to modern man. But, how do we, as Christians, tackle this ethical dilemma? Are we playing God when we use Biotechnologies to manipulate life? Listen to this interview by Alexa Ho more Bioethics
The Journey to Emmaus or Spiritual Direction on the Walk
Two men were walking towards the town of Emmaus, about seven miles from Jerusalem. They were discussing animatedly about the events of Jesus’ claims, his death, and reports of his resurrection when a third man joined them. This man explained the significance of the events through the Scriptures. During the evening meal at Emmaus, the two men recognized the third man as Jesus! (Luke 24:13-33). They were enlightened both by the dialogue and Jesus’ explanation of the Scriptures until they feel their hearts burning within them. The Truth turned their despair to joy when they beheld the risen Christ. read more more on Spiritual Direction
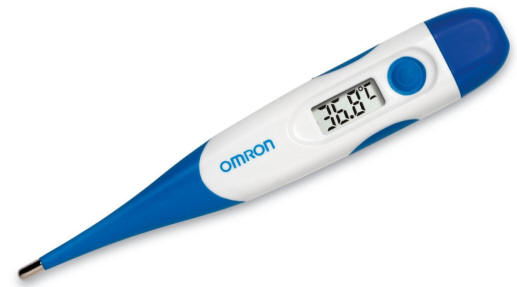
What is your temperature? A spiritual thermometer
Wang Ming Tao, a famous Chinese Christian taught us to always check our life with a spiritual thermometer so that we will be spiritually healthy when the Lord comes. Read more More e-Reflections
Writing is hard and gruelling work. It is incubated amidst blood, sweat, and tears. No, these aren’t the ink I write with. I just want to express that it not just physically challenging, but mentally too. The thought of all the work frightens me, as it involves transferring all those ideas that I carry with me in my mind, into words. Ideas brew and form, and run ahead of writing—writing about them is akin to playing ‘catch up’. Such imagery is enough to make me feel breathless. Writing, especially in an authentic voice, makes me exceptionally vulnerable, as my inner thoughts and aspirations are made bare to the world. Such glaring spotlight is not easy on me—an extreme introvert. read more more A Writing Life
Read more more Poems
A wonderful painting of the scene at Emmaus where the two travellers recognised Jesus as the Resurrected Christ (Luke 24:13-32) is a favourite theme of Rembrandt. He frequently returned to this scene in his sketches and paintings. This oil painting titled The Supper at Emmaus completed in 1648 during his so-called ‘mature’ years was in complete contrast with another painting one the same theme that was done during his ‘younger’ years.
read more
more e-Reflections
Waiting for the Light: Mastery Inactivity
This is often what happens when we are hit with some catastrophes in our lives. In such situation, we are full of an urgency to act. An urgency to do something to get us out of the situation. Anything at all, even though the action may not be beneficial or at times may cause harm. An alternative option is to sit idly by and ride out the storm.
read more
more e-Reflections
Delphi, the center of the world
Delphi is an interesting place to visit in Greece. It is famed for its Oracle and being the centre of the ancient Greek world. There are numerous legends as to its origin. One is that of Apollo. Apollo when he was only an infant shot an arrow which killed Python “a dragon”. Python was the son of Gaia (mother earth or goddess). Apollo had to atone for this by suffering a period of menial labour. Python was guarding the navel of the world. The location was Delphi. read more more on Bible Lands
The Biblical and Theological Foundations of Spiritual Formation
Christian spiritual formation is a process grounded on the biblical and theological concepts of restoration, relationship, and shalom. These key foundational concepts are as follows: 1. Restoring the imago Dei 2. Relationship with the triune God 3. Shalom and the kingdom of God read moremore Spiritual Formation
The Nature of Spiritual Formation
Writing to the Christians in Corinth about spiritual transformation (2 Corinthians 3:18), Paul notes that “we, who with unveiled faces all reflect the Lord’s glory, are being transformed into his likeness with ever-increasing glory, which comes from the Lord, who is the Spirit.” It is Paul’s intention to emphasize (1) that in spiritual transformation Christians (individuals and the Christian faith community) will be transformed into a likeness of Christ, (2) that this transformation is an ongoing process, (3) that it is Trinitarian, (4) that the Holy Spirit is involved in this transformation, and (5) that God’s glory is thereby restored. read more more Spiritual Formation
|
|
Dear friends, Welcome! Come on in, make yourself a cup of coffee and let your mouse do the clicking. Look around and may you find rest for your body , nourishment for your mind and refreshment for your soul. Join me in a continuing dialogue on everyday Christian spirituality, spiritual formation and transformation, spiritual direction, theology, Christian education, biomedical ethics, Star Trek, postmodern parenting and science fiction. I enjoy reading and watching movies. I write and preach in churches, speak in conferences and lead retreats. My other vocation is as a paediatrician and a medical educator.
Connect with me
Blogs
Spiritual Formation on the Run
spirituality, culture and life
I value your comments and your dialogue
Tweets by @alexthtang
|





















.png)





.jpeg)





.JPG)
























updated 05-Jan-24 01:49:25 PM